
 ^^^
^^^
Group of Structure and Function of Proteins and Computational Biology

Senior Scientist
Dr. David Řeha, PhD
Scientist
Daniel Bonhenry, Ph.D.
Associated Scientist
Ing. Eva Cséfalvay, PhD.
Dr. Natallia Kulik, PhD.
Graduate Student
MSc. Zeenat Zara
Staff
Magda Vojtová
Mgr. David Řeha, PhD
Phone: +420 389 033 801
reha@nh.cas.cz
Address
Zámek 136, CZ 373 33 Nové Hrady
Research
The Laboratory of Structure and Function of Proteins was founded in 2002 as a joint laboratory of the Institute of Systems Biology and Ecology of the Academy of Sciences and the Institute of Physical Biology of the University of South Bohemia named Laboratory of High Performance Computing . The fundamental methods were the demanding computational techniques that cover a range of processes involving solving structure, interactions and dynamics of proteins and membranes up to the conformational changes of small molecules such as nucleic acids and carbohydrates. The relationship between the structure and function of the proteins and their complexes became a central research issue. Continuesly growing, the laboratory today combines methods ranging from computational and spectroscopic, to molecular biological, biochemical, and protein crystallisation. With its focus on molecular systems biology, the relationship between structure and function of proteins, dynamic changes related to functional processes on the level of proteins and the mutual interaction of cofactors and subunits in protein complexes the laboratory is an integral part of the new research concept of the Institute. The research approach is very complex using various methods of protein research with a synthesis of theoretical and experimental methods. Molecular dynamics, quantum chemical and semiempiric calculations of optical spectra, charge localisation or energy transfer and molecular modelling methods are combined with mostly spectroscopic and crystallographic methods and methods of protein structure determination. The interaction of biomolecules with ligands (drugs) is also studied using QM/MM calculations and molecular docking. The laboratory also studies the mechanisms of enzymatic reactions using QM/MM calculations.The laboratory has highly ambitious aims with regard to the development of new methods in molecular systems biology and its application to systems of a common interest, hot topic systems.
Modulation of human ORAI1 channels: modeling and simulations
Researchers involved from the group: Daniel Bonhenry, Saurabh Kumar Pandey, Deepti Mishra, David Reha, Rudiger H. EttrichExternal collaboration: Irene Frischauf, Isabella Derler, , Christoph Romanin (Johannes Kepler University Linz, Austria) and Rainer Schindl (Medical University of Graz, Austria)
Orai1 is a calcium-selective channel located in the plasma membrane, and belongs to the family of calcium release activated channels (CRAC). Orai1, as component of store-operated calcium entry (SOCE), is activated by the second component of SOCE, STIM1, when intracellular calcium stores are depleted. STIM1, located at the endoplasmatic reticulum (ER), senses levels of calcium in the ER and is activated by calcium store depletion. In turn, calcium influx via Orai1 channel refills calcium levels in the endoplasmic reticulum. Based on the Drosophila melanogaster Orai crystal structure a homology model of human Orai1was prepared in our group that includes extracellular and intracellular loops existing only in the human isoform. The sequence and architecture of Orai channels is unique among other ion channels and suggests a novel gating mechanism. The selectivity filter is formed by a ring of six glutamate residues followed by a hydrophobic and consequent basic region further down the pore. The pore extends into cytosol by approximately 20 Å. Using combined experimental and theoretical approaches this study focuses on the central ion pore to investigate the gating mechanism of this unique channel including altered gating of Orai1 mutants occurring in tumor cells, the communication between the intracellular loop and the N-terminus, or its interaction with STIM1.
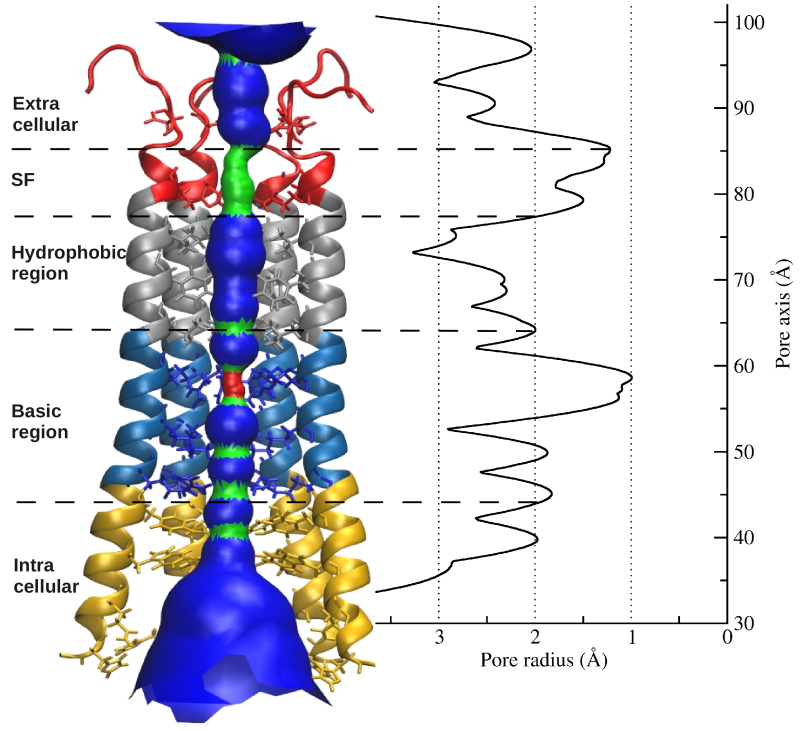 Picture: human Orai1 pore radius
Picture: human Orai1 pore radius
Recent Publications from our group
- [1] Irene Frischauf, Vasilina Zayats, Michael Deix, Anna Hochreiter, Isaac Jardin Polo, Martin Muik, Barbara Lackner, Barbora Svobodova, Teresa Pammer, Monika Litvinukova, Amrutha Arumbakam Sridhar, Isabella Derler, Ivan Bogeski, Christoph Romanin, Rüdiger Ettrich*, Rainer Schindl* (2015) A calcium accumulating region, CAR, in the Orai1 channel enhances Ca2+- permeation and SOCE-induced gene transcription Science Signaling 8 (408): ra131 (Cover story)
- [2] Derler, Isaac Jardin, Peter B Stathopulos, Martin Muik, Marc Fahrner,Vaselina Zayats, Saurabh Pandey, Michael Poteser, Barbara Lackner, Marketa Absolonova, Klaus Groschner, Rüdiger Ettrich, Ikura, Christoph Romanin (2016) Cholesterol binding modulates Orai1 channel function Science Signaling 9 (412): ra10. Jan
- [3] Irene Frischauf, Monika Litviňuková, Romana Schober, Vasilina Zayats, Barbora Svobodová, Daniel Bonhenry, Victoria Lunz, Sabrina Cappello, Laura Tociu, David Reha, Amrutha Stallinger, Anna Hochreiter, Teresa Pammer, Carmen Butorac, Martin Muik, Ivan Bogeski, Rüdiger Ettrich, Christoph Romanin, Rainer Schindl (2017) Transmembrane helix connectivity in Orai1 controls two gates for calcium-dependent transcription Science Signaling 10 (507): eaao0358. (Cover story)
- [4] Marc Fahrner, Saurabh K Pandey, Martin Muik, Lukas Traxler, Carmen Butorac, Michael Stadlbauer, Vasilina Zayats, Adéla Krizova, Peter Plenk, Irene Frischauf, Rainer Schindl, Hermann J Gruber, Peter Hinterdorfer, Rüdiger H Ettrich, Christoph Romanin, Isabella Derler (2018) Communication between N-terminus and Loop2 tunes Orai activation The Journal of Biological Chemistry 293: 1271-1285.
Skeletal Bone Deformity: Role of PAPS-Synthase in Sulfur Metabolism and the Related Dysplasia/Diseases
Researchers involved from the group: , David Reha, Magda Vojtova, Rudiger H. Ettrich,External collaboration: K.V. Venkatachalam (College of Medical Sciences, Nova Southeastern University, Ft. Lauderdale, FL, USA ) and Dhiraj Sinha (CNRS Marseille , France)
Bone deformations and skeletal dysplasias are relatively rare, but collectively the birth incidence of these disorders is almost 1/5000 (Orioli IM, Castilla EE, Barbosa-Neto JG, The birth prevalence rates for the skeletal dysplasias, J Med Genet 1986;23:328–332.). These disorders range in severity and can be associated with a variety of orthopedic, neurologic, auditory, visual, pulmonary, cardiac, renal, and psychological complications. More than 350 skeletal dysplasias have been characterized to date primarily on the basis of clinical, radiographic, and molecular criteria. (Superti-Furga A, Unger S., Nosology and classification of genetic skeletal disorders: 2006 revision. Am J Med Genet A 2007;143:1–18). They all have in common that they not only result from mutations in various families of genes that encode extracellular matrix proteins, but also transcription factors, tumor suppressors, signal transducers (ligands, receptors, and channel proteins), enzymes, cellular transporters, chaperones, intracellular binding proteins, RNA processing molecules, cilia and cytoplasmic proteins, and a number of gene products of currently unknown functions. One of the skeletal deformity for example is described as Spondylo Epi Metaphyseal Dysplasia (SEMD), which results from mutations in genes that are linked to the sulfonation pathway that encodes 3’-phosphoadenosine 5’-phosphosulfate Synthase (PAPSS). Inorganic sulfate, though important for life, is biochemically inert and cannot be used by the cell directly. Inorganic sulfate is activated to 3’-phosphoadenosine 5’-phosphosulfate (PAPS), a sulfur nucleotide, which is the universal sulfuryl donor of the cell. In mammals 3’- phosphoadenosine 5’-phosphosulfate Synthase (PAPSS), using ATP, converts biochemically inert inorganic sulfate to the metabolically active PAPS. It is a bi-functional enzyme and catalyzes the formation of PAPS in two sequential steps. In the first step, inorganic sulfate reacts with ATP to form APS and pyrophosphate. The resulting phosphoric-sulfuric anhydride bond has high energy that is the chemical basis of sulfate activation. The second step is catalyzed by the kinase domain of PAPSS and involves the reaction of APS with ATP to form PAPS and ADP. The proper function of PAPSS is essential for normal physiology in the human being. Although its overall mechanism and kinetics have been well studied in the past, more recent discoveries including the resolution of its crystal structure and research in its regulatory functions revealed previously unanticipated behaviors. As the ubiquitous sulfate donor in most biological systems, the product of the enzyme, PAPS, plays an essential role in ECM formation, embryonic development and biomolecule secretion. Final results will describe the nature of the PAPSS dynamics and its intermolecular interactions including ligands. Using smart combinations of experimental (kinetics, crystallography, biochemistry) and computational (classical molecular dynamics (MD), docking, interaction energies, and hybrid QM/MM calculation) methods, we try to understand how in WT as well as in disease relevant mutations the three dimensional structure of PAPSS determines the enzyme function, focusing on the roles of specific amino acid residues/overall structures on the dynamics of the enzyme in aqueous solution and the related quaternary arrangements of the enzyme. Enzymatic reactions are predicted/described in three-dimensional space and the reaction coordinate is explored through the lens of molecular dynamics simulations, hybrid QM/MM and quantum calculations. Results are discussed that give a realistic picture of the enzyme activity including molecular interactions, transition state structures and the reaction coordinate. The novel aspect of our study will be that for the first time, direct structural consequences of the functionally critical mutations will be described at a molecular level. This includes, altered dynamics and ligand binding properties which are the prerequisites for future studies targeted towards specific drug design.
Recent Publications from our group
- [1] Lydie Plackova, Dhiraj Sinha, Chris Soha, David Reha, Rudiger H. Ettrich, K.V. Venkatachalam (2018) The connecting link: Role of the linker region on the ATPsulfurylase and APSkinase structure-function of human PAPS-Synthase, prepared for submission the The Biophysical Journal in 2/2018
Interdomain communication in Type I restriction-modification enzymes
Researchers involved from the group: Eva Csefalvay, Pavel Grinkevich, David Reha, Rudiger H. EttrichInternal collaboration: Ivana Kuta Smatanova, Katja Shamayevova, Marie Weiserova
External collaboration: Jannette Carey (Princeton University, NJ, USA) and Dhiraj Sinha (CNRS Marseille , France)
Type I restriction-modification enzymes are multifunctional heteromeric complexes with DNA cleavage and ATP-dependent DNA translocation activities located on motor subunit HsdR. Functional coupling of DNA cleavage and translocation is a hallmark of the Type I restriction systems that is consistent with their proposed role in horizontal gene transfer. DNA cleavage occurs at nonspecific sites distant from the cognate recognition sequence, apparently triggered by stalled translocation. The first X-ray crystal structure of the complete HsdR subunit from E. coli plasmid R124 was reported by our lab and suggested that the triggering mechanism involves interdomain contacts mediated by ATP. In vivo and in vitro activity assays of WT and mutant protein are combined with computational modeling to probe interdomain contacts potentially involved in triggering DNA cleavage that occurs at nonspecific sites distant from the cognate recognition sequence. Analysis of the computational modeling allows to assign specific roles for the individual domains in either translocation or restriction and predicts interdomain engagement responsible for signal transmission between the endonuclease and helicase domains and interdomain contacts involved in global conformational changes connected to the enzymatic function. The combined experimental and modeling results so far suggest that the ATP ligation state of the enzyme might be communicated through the endonuclease/motor subunit by a cascade of linked local and global conformational changes. Such large-scale domain motions are expected to be readily detected by the other enzyme subunits, and may represent a signal reporting the ATP ligation state of the endonuclease/motor subunit. Current focus is on the role of a newly detected 5th domain of the motor subunit and its role in complex formation with the Methyltransferase and/or DNA binding.
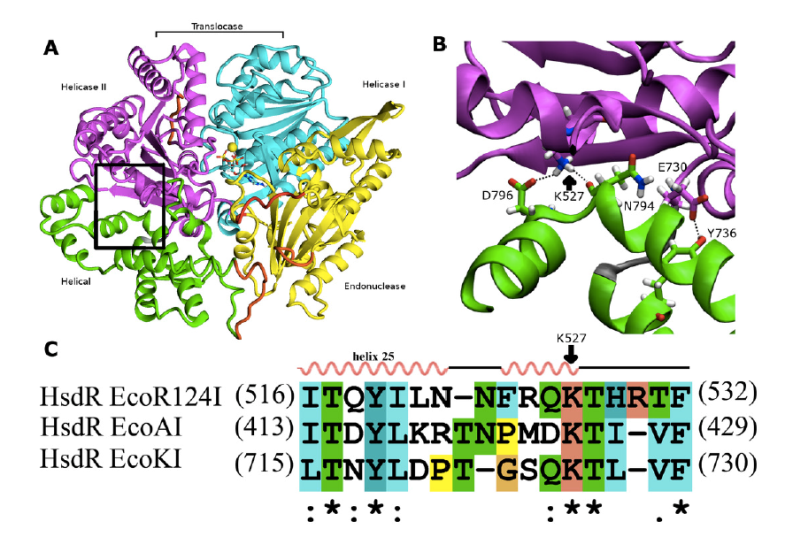 Picture: Interdomain interactions at the helical-helicase 2 domain interface in the HsdR subunit of EcoR124I restriction-modification complex
Picture: Interdomain interactions at the helical-helicase 2 domain interface in the HsdR subunit of EcoR124I restriction-modification complex
Recent Publications from our group
- Mikalai Lapkouski, Santosh Panjikar, Pavel Janscak, Ivana Kuta Smatanova, Jannette Carey, Rüdiger Ettrich*, Eva Csefalvay (2009) Structure of the motor subunit of type I restriction-modification complex EcoR124I. Nat Struct Mol Biol 16: 1. 94-95
- Eva Csefalvay, Mikalai Lapkouski, Alena Guzanova, Ladislav Csefalvay, Tatjana Baikova, Igor Shevelev, Vitali Bialevich, Katsiaryna Shamayeva, Pavel Janscak, Ivana Kuta Smatanova, Santosh Panjikar, Jannette Carey, Marie Weiserova, Rüdiger Ettrich* (2015) Functional coupling of duplex translocation to DNA cleavage in a type I restriction enzyme PLOS One, PLoS ONE 10 (6): e0128700
- D Sinha, K Shamayeva, V Ramasubramani, D Reha, V Bialevich, M Khabiri, N Milbar, A Guzanova, M Weiserova, E Csefalvay, J Carey, R Ettrich* (2014) Interdomain communication in the endonuclease/motor subunit of Type I restriction-modification enzyme EcoR124I Journal of Molecular Modeling 20 (7): 2334.
- Pavel Grinkevich, Iuliia Iermak, Nicholas A Luedtke, Jeroen R Mesters, Rudiger Ettrich*, Jost Ludwig (2016) pHluorin(GFP)-assisted expression, purification, crystallization and X-ray diffraction data analysis of C-terminal domain of HsdR subunit from E. coli type I restriction-modification system EcoR124I. Acta Crystallographica Section F, 72 (9): 672- 676.
- Bialevich V, Sinha D, Shamayeva K, Reha D, Guzanova A, Csefalvay E, Carey J, Weiserova M and Ettrich R.* (2017) The helical domain of the motor subunit of EcoR124I participates in ATPase activity and dsDNA translocation, PeerJ, Volume 2017, Issue 1, 2017, Article number e2887.
- Dhiraj Sinha, Vitali Bialevich, Katsiaryna Shamayeva, Alena Guzanova, Alexandra Sisakova, Eva Csefalvay, Lumir Krejci, Jannette Carey, Marie Weiserova, Rüdiger H Ettrich* (2018) A residue of motif III positions the helicase domains of motor subunit HsdR in restriction-modification enzyme EcoR124I, Journal of Molecular Modeling, invited paper for the topical issue “Modelling Interactions in Biomolecules VIII” (submitted January 31, 2018).
Computational modeling of domain motion of Topoisomerase IA during enzyme bridging strand-passage mechanism.
Researchers involved from the group: Natalia Kulik, Daniel Bonhenry, Rudiger H. Ettrich, David RehaExternal collaboration: Yuk-Ching Tse-Dinh (Biomolecular Sciences Institute of Florida International University, Miami, USA) and Dhiraj Sinha (CNRS Marseille , France)
Eukaryotic topoisomerases I are the targets of an increasing number of anti-cancer and anti-tumor drugs that act by inhibition these enzymes. Topoisomerase IA from E.coli is an enzyme, involved in relaxation of negatively supercoiled DNA, to preserve correct DNA topology during replication and transcription. Cleavage and religation of DNA during catalysis is accompanied by large conformational changes of the enzyme-DNA complex. Available crystal structures might represent different steps of the catalytic cycle, although details of domain interaction and loop motion are still unknown. The project focuses on computationally modeling the individual conformations corresponding to specific steps of the catalytic cycle for the full-length enzyme, on the describtion of a dynamic model of the catalytic process and on revealing interactions within the enzyme-DNA complex. It aims to predict alternative ways for inhibition of DNA replication in bacteria cells. This project represents a close collaboration with Prof. Yuk-Ching Tse-Dinh, Ph.D at the Biomolecular Sciences Institute of Florida International University, Miami, and involves docking of potential inhibitors in the active site of topoisomerase IA from E.coli, study of interactions and validation. Computational methods like molecular docking, molecular dynamics simulation and quantum mechanics methods will focus on the prediction of potential antimicrobial drugs, potentially lowering the experimental costs and time. In a first step possible binding places for binding of different drugs are identified and prominent inhibitors are selected, predicting possible effects on enzyme action. Several approaches are used for searching and analysis of inhibitors, including characterization geometry of binding partners (Yasara, VMD), calculation of energy parameters such as binding affinity and charge distribution (AutoDock, Schrodinger) and determination of transition state conformations with semi- empirical and quantum approaches.
Was binding of free amino acids an early innovation in the evolution of allostery?
Researchers involved from the group: Saurabh Kumar Pandey, Rudiger H. Ettrich, David RehaInternal collaboration: Babak Minofar
External collaboration: Jannette Carey (Princeton University, NJ, USA), Milan Melichericik (Comenius University, Bratislava, SK) and Thomas Stockner (Medical University Vienna, Austria)
Interpretation of thermodynamic ligand-binding data through the lens of molecular dynamics has led to a structural and energetic description of the molecular mechanism of allostery for the hexameric E. coli arginine repressor, the master feedback regulator of transcription in L-arginine metabolism, which displays strong negative cooperativity of L-arginine binding. A controversial prediction of the famous allostery model of Monod, Wyman, and Changeux is that constraints imposed on protein subunits by multimerization are relaxed by ligand binding, but with conservation of symmetry in partially-liganded states. The ~50 kDa hexameric C-terminal domain was studied by 100 ns molecular dynamics simulations in the presence and absence of the six L-arg ligands that bind at the trimer-trimer interface. A rotational shift between trimers followed by rotational oscillation occurs in the production phase of the simulations only when L-arg is absent. Analysis of the system reveals that the degree of rotation is correlated with the number of hydrogen bonds across the trimer interface. The trajectory presents frames with one or more apparently open binding sites into which one L-arg could be docked . The simulations reveal that conserved Arg and Asp sidechains in each L-arginine binding pocket promote rotational oscillation of apoArgR trimers by engagement and release of salt bridges. Binding of exogenous L-arginine displaces resident Arg residues and arrests oscillation, shifting the equilibrium quaternary ensemble and promoting subunit motions that generate an entropic driving force while maintaining symmetry in partially-liganded states. The results indicate that partially-liganded states can be structurally symmetric despite their conceptual asymmetry. The symmetric relaxed state is visualized as a multimer with all subunits anchored near the center, and with motions transferred to the periphery like a bouquet of balloons in strong wind. Thus, even during sequential filling of binding sites, symmetry can be maintained by exploiting the dynamics of the assembly and the distributed nature of its cohesive energy. The mechanism suggests the possibility that binding of free amino acids was an early innovation in the evolution of allostery. Current research is focused on elucidating the detailed mechanism in intact, full-length protein in the various branches of the Arg-Repressor family and thus understanding how Arg-Repressor evolution might have occurred.
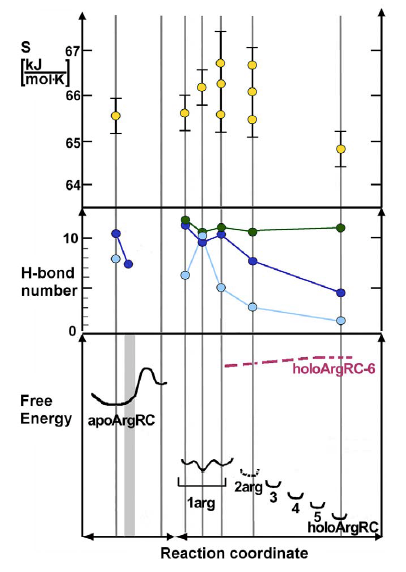 Picture: Reaction coordinate for L-Arginine binding (6x L-Arg) to Arg-Repressor from E. coli. Vertical lines mark coordinates for which values of conformational entropy, inter-trimer hydrogen bond number, and relative free energy are plotted based on inspection or calculation from simulation
Picture: Reaction coordinate for L-Arginine binding (6x L-Arg) to Arg-Repressor from E. coli. Vertical lines mark coordinates for which values of conformational entropy, inter-trimer hydrogen bond number, and relative free energy are plotted based on inspection or calculation from simulation
Recent Publications from our group
- Rebecca Strawn, Milan Melichercik, Michael Green, Thomas Stockner, Jannette Carey, Rudiger Ettrich* (2010) Symmetric allosteric mechanism of hexameric Escherichia coli arginine repressor exploits competition between L-arginine ligands and resident arginine residues PLOS Computational Biology 6: 6. e1000801
- R Strawn, T Stockner, M Melichercik, L Jin, W - F Xue, J Carey, R Ettrich* (2011) CHAPTER FIVE: Synergy of molecular dynamics and isothermal titration calorimetry in studies of allostery. In: Methods in Enzymology, Biothermodynamics, Part D Volume 492 Edited by:Michael L. Johnson, Jo M. Holt , Gary K. Ackers. pages 151-189 Academic Press - Elsevier Publishing Company, ISBN: 978-0-12-386003-3
- S K Pandey, D Reha, V Zayats, M Melichercik, J Carey, R Ettrich *(2014) Bindingcompetent states for L-arginine in E. coli arginine repressor apoprotein. Journal of Molecular Modeling 20 (7): 2330.
Structure and function of β-N-acetylhexosaminidases and their inhibition via new, non-natural substrates
Researchers involved from the group: Natallia Kulik, Rudiger H. EttrichInternal collaboration: Kristyna Slamova, Vladimir Kren
β-N-Acetylhexosaminidases (β-Hex) (2-acetamido-2-deoxy-β-D-hexopyranoside acetamido-deoxyhexohydrolases, EC 3.2.1.52.; CAZy GH20 and 84 - Carbohydrate Active Enzymes, http://www.cazy.org/) are exo-glycosyl hydrolases, catalyzing the cleavage of terminal β-D-GlcNAc and β-D-GalNAc1 residues. Dysfunctions in human hexosaminidases (GH20) result in serious lysosomal storage disorders (Sandhoff and Tay-Sachs diseases), which draws attention to the structure and catalytic mechanism of these enzymes. Fungal β-N-acetylhexosaminidases are known to be instrumental in the chitinolytic system in the fungal cell wall. The so-called “tandem binary system”, produced under a severe regime of catabolite repression, consists of chitinases and β-N-acetylhexosaminidases and is responsible for the control of cell wall chitin lysis. Some mycoparasitic fungi produce β-Hex upon specific induction by chitin or fungal cell walls, thus serving as potentially useful biocontrol agents against plant pathogens.
Small molecule inhibitors of family GH20 and 84 enzymes are very important both as tools for elucidating the role of these enzymes in biological processes as well as for developing therapeutic interventions with minimal side effects. A major obstacle in the generation of selective inhibitors of human hexosaminidases is a presence of functionally related enzyme O-GlcNAcase in the organism. Thus, the main goal is now to design highly selective inhibitors of human lysosomal β-Hex and nucleocytoplasmic O-GlcNAcase. The promiscuity of inhibitors and the resulting inhibition of both types of GlcNAc-cleaving enzymes in vivo necessarily generates complex phenotypes, which limits their utility in studying the physiological roles of individual enzymes. Such unspecific inhibitor cannot be used as therapeutics as it would cause cell damage and a death.
A broad range of computational methods is applied, ranging from homoloy modeling to quantum calculations, and results are confronted with experimental data gained by the internal collaborators in Prague.
Recent Publications from our group
- Kristýna Slámová, Natallia Kulik, Martin Fiala, Jana Krejzová-Hofmeisterová, Rüdiger Ettrich, Vladimír Křen (2014) Expression, characterization and homology modeling of a novel eukaryotic GH84 β-N-acetylglucosaminidase from Penicillium chrysogenum Protein Expression and Purification 95: 204–210.
- Jana Krejzová, Kristýna Slámová, Petr Šimona, Lubica Kalachova, Natallia Kulik, Pavla Bojarová, Petr Marhol, Helena Pelantová, Josef Cvačka, Rüdiger Ettrich, Vladimír Křen (2014) Inhibition of GlcNAc-processing glycosidases by C-6-azido-NAG-thiazoline and its derivatives Molecules 19: 3471-3488.
- Kristýna Slámová, Radek Gažák, Pavla Bojarová, Natallia Kulik, Rudiger Ettrich, Helena Pelantová, Petr Sedmera, Vladimír Křen (2010) 4-Deoxy-substrates for beta-N acetylhexosaminidases: how to make use of their loose specificity Glycobiology 20: 8. 1002- 1009.
- Natallia Kulik, Kristyna Slamova, Rudiger Ettrich, Vladimir Kren (2015) Computational study of β-N-acetylhexosaminidase from Talaromyces flavus, a glycosidase with high substrate flexibility BMC Bioinformatics 16 (1): 28.
- Natallia Kulik, Lenka Weignerová, Tomáš Filipi, Petr Pompach, Petr Novák, Hynek Mrázek, Kristýna Slámová, Karel Bezouška, Vladimír Křen, Rüdiger Ettrich* (2010) The α-galactosidase type A gene aglA from Aspergillus niger encodes a fully functional α-Nacetylgalactosaminidase Glycobiology 20: 11. 1410-1419.
Former PhD students
Saurabh Pandey (PhD., 2018), now post-doc in our group
Pavel Grinkevich (PhD., 2018), now University of South Bohemia in Ceske Budejovice
Vitali Bialevich, Ph.D (PhD., 2017), now Centre Algatech, Trebon
Dhiraj Sinha (PhD., 2017)
Katsiarina Shamayeva (PhD., 2015), now post-doc in group of Membrane Physiology and Bioenergetics
Vasilina Zayats (PhD., 2014), now University of Warsaw, Poland
Iryna Kishko (PhD., 2013)
Morteza Khabiri (PhD., 2011), now University of Michigan, Ann Arbor
Natalia Kulik (PhD., 2011), now staff scientist in our group
Abdul Samad (PhD., 2010), Institute of Microbiology, Prague
Jaroslava Kohoutova (PhD., 2010), now University of South Bohemia
Mikalai Lapkouski (PhD., 2008), now Karolinska Institute
Peter Palencar (PhD., 2007), now Slovak Academy of Sciences
Links
Recent international collaborations:
Dr. Alex de Vries, Groningen University, Netherlands
Dr. Christoph Romanin, Johanner Kepler University, LInz, Austria
Dr. Jost Ludwig, Bonn University, Germany
Prof. Stephan Grissmer, Ulm University, Germany
Dr. Thomas Stockner, Vienna Medical University, Austria
Prof. Jannette Carey, Chemistry Department, Princeton University, NJ, USA
Dr. Santosh Panjikar, EMBL outstation, DESY, Hamburg, Germany
Dr. Juan B. Arellano, Instituto de Recursos Naturales y Agrobiologia de Salamanca (IRNASA–CSIC), Salamanca, Spain
Dr. Pavel Janscak, Institute of Molecular Cancer Research, University of Zürich, Zürich, Switzerland
Prof. Norbert Mueller, Johannes Kepler University, Linz, Austria
